Apitegromab for lean mass preservation during tirzepatide-induced weight loss: a randomized, double-blind, placebo-controlled phase 2 trial
Trial details
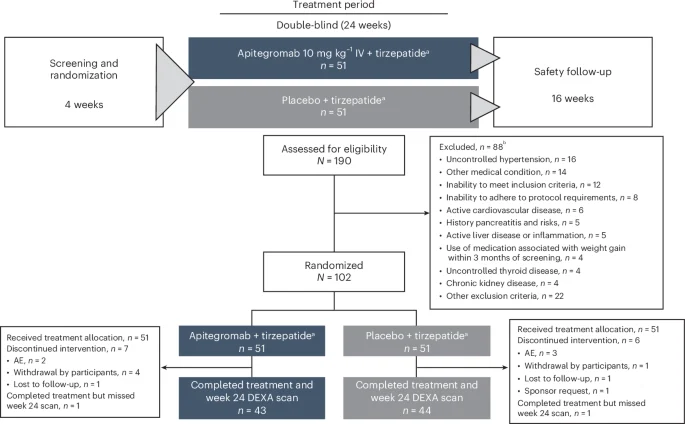
EMBRAZE was a randomized, double-blind, placebo-controlled, phase 2 trial (NCT06445075) conducted at seven sites in the United States to evaluate the efficacy, safety, pharmacokinetics and pharmacodynamics of apitegromab in adults receiving tirzepatide. The trial protocol was approved by Advarra, a central institutional review board (IRB), as well as IRBs at participating sites and adhered to the Declaration of Helsinki and Good Clinical Practice guidelines. IRBs at participating sites included ProSciento CRU (Chula Vista, California); AdventHealth Translational Research Institute (Orlando, Florida); Great Lakes Clinical Trials, LLC, dba Flourish Research (Chicago, Illinois); Tandem Clinical Research GI, LLC (Marrero, Louisiana); Alliance for Multispecialty Research, LLC (Norman, Oklahoma); Apex Mobile Clinical Research (Bellaire, Texas); and Clinical Trials of Texas, LLC, dba Flourish Research (San Antonio, Texas). The study protocol was amended from version 1 to version 2 prior to enrollment of the first participant to add tirzepatide as an incretin mimetic therapy and to designate AEs of special interest (pancreatitis, liver injury, depression/suicidality and creatine kinase elevations).
Participants
Eligible participants were aged 18−65 years with either a BMI of ≥27.0 to −2 and at least one weight-related comorbid condition or a BMI of ≥30.0 to ≤45 kg m−2. Stable body weight (±5 kg) within 90 days of screening and at least one self-reported unsuccessful dietary effort to lose weight were also required. Females of childbearing potential were required to have a negative pregnancy test and agree to use adequate birth control throughout trial participation. In addition, participants were required to adhere to the study visit schedule and the prespecified prohibitions and restrictions (Study Protocol).
Individuals with a history of or active cardiovascular disease were ineligible, including clinically significant arrhythmias; congestive heart failure (New York Heart Association grades I−IV); mild, moderate or severe coronary artery disease or history of angina; uncontrolled hyperlipidemia; myocardial infarction, stroke, coronary artery bypass graft surgery or percutaneous coronary intervention; valve disorders or defects; abdominal aortic aneurysm; or pulmonary hypertension. Also excluded were individuals with uncontrolled hypertension (American Heart Association stage 1 or stage 2) or history of hypertensive crisis or treatment-resistant hypertension; history of or active ischemic, hemorrhagic or anatomical neurovascular disease, including transient ischemic attack, cerebrovascular accident, arteriovenous malformation or brain aneurysm, peripheral vascular disease such as deep vein thrombosis/pulmonary embolism, chronic venous insufficiency, claudication or lymphedema; active pulmonary disease, including chronic obstructive pulmonary disease, pulmonary fibrosis, moderate-to-severe sleep apnea and moderate-to-severe asthma; hepatic, pancreatic, neuromuscular and/or psychiatric disease; active malignancy (other than local subcutaneous squamous cell and basal cell carcinomas); or history of immunosuppressive, chemotherapeutic or radiation treatment within 12 months prior to screening. Individuals with a history of type 1 or type 2 diabetes were also excluded. Type 2 diabetes that was resolved for more than 12 months and prediabetes managed with lifestyle and diet were not exclusions. Additional exclusion criteria included history of gastroparesis, gastric or peptic ulcer, active gastritis or esophagitis, uncontrolled gastroesophageal reflux disease or severe inflammatory bowel disease; uncontrolled thyroid disease; severe endocrine disorders such as Cushingʼs disease, hypogonadism and growth hormone deficiency; autoimmune or inflammatory disorders that may cause muscle wasting, such as myasthenia gravis, rheumatoid arthritis, lupus, polymyositis or dermatomyositis; neuromuscular disorders that may cause muscle wasting, such as muscular dystrophy, spinal muscular atrophy or amyotrophic lateral sclerosis; neurologic diseases such as epilepsy, dementia, Parkinsonʼs disease or Bellʼs palsy; acute or chronic pancreatitis (or at high risk for pancreatitis), clinically significant abnormal lipase and/or amylase or taking medications that may cause serious damage to the pancreas, such as valproic acid; history of or active acute or chronic liver disease; uncontrolled psychiatric disease; severe coagulopathy; chronic kidney disease stages 1−5; any chronic active infection or treatment of infection within 6 months prior to screening; bariatric surgery or use of gastric balloons or other gastric volume reduction devices; or donation or loss of ≥500 ml (1 pint) of blood within 8 weeks prior to screening or plasma donation of ≥600 ml within 14 days prior to screening.
Individuals with a history of apitegromab treatment were ineligible. Other medication-related exclusions included use of antiobesity medications, nutritional supplements or over-the-counter products for weight loss within 3 months prior to screening; use of antidiabetic medications, nutritional supplements or over-the-counter products for lowering blood sugar within 3 months prior to screening; use of medications known to induce weight gain within 3 months prior to screening; use of therapies with potentially significant muscle effects (for example, androgens, insulin-like growth factor, growth hormone, systemic beta-agonists, neurotoxins or muscle relaxants or muscle-enhancing supplements) in any form within 3 months prior to screening; use of systemic corticosteroids within 60 days before screening or intraarticular corticosteroid injection (inhaled or topical steroids were allowed); or medications that impede coagulation or platelet aggregation. History of alcoholism or illicit drug use or the use of inhaled vasoconstrictive tobacco or cannabis or synthetic products, such as vape pens, pipes, cigars and cigarettes, were prohibited. Individuals were also ineligible if they had any contraindications to study treatment or if, in the opinion of the investigator, they had any other medical condition, clinically significant laboratory result or electrocardiogram findings that may compromise safety or compliance, preclude the participant from successful study completion or interfere with interpretation of results.
Procedures
Baseline characteristics were collected via standard assessments and a complete physical examination (see Study Protocol for additional details). Demographic information, such as age, sex, race and ethnicity, were self-reported by eligible participants. Participant sex was not considered in the study design; however, it was used as a covariate in the data analysis as described in the statistical methods section, and post hoc subgroup analysis of the primary endpoint by sex was performed on an exploratory basis. All participants received standard care counseling with regard to lifestyle recommendations such as diet, physical activity and behavior modification; however, reporting on these variables was not mandated or monitored.
Participants were randomized (1:1 via an interactive web response system managed by an independent vendor, without blocking or stratification factors) to receive apitegromab plus incretin mimetic therapy or placebo plus incretin mimetic therapy. The study sponsor, study participants, investigators and site personnel, with the exception of the pharmacist, were blinded to treatment assignments. The site pharmacist remained unblinded throughout the duration of the trial. DEXA scans were performed and assessed by blinded personnel, and statistical analyses were planned prior to unblinding at the end of the treatment period.
Apitegromab and placebo treatments were administered intravenously. After a 4-week screening period and written informed consent, site personnel administered an initial dose of tirzepatide 2.5 mg plus apitegromab or placebo (day 1). Thereafter, participants were trained to administer weekly doses of tirzepatide using an injection pen for 24 weeks. Tirzepatide dose was increased by 2.5 mg every 4 weeks according to the recommended dose-escalation schedule20 and subject to treatment response and tolerability up to a maximum maintenance dose of 15 mg per week. No participants received semaglutide as a treatment assignment, due to a lack of drug availability at the time of EMBRAZE study initiation, which lasted throughout study duration. Participants received apitegromab or placebo every 4 weeks for 24 weeks, followed by a 16-week safety period during which study treatments were not administered.
Concomitant therapies or interventional procedures medically indicated as part of standard supportive care for the participant were permitted at the discretion of the investigator. Antiobesity medications, antidiabetic medications or medications associated with serious risk to the pancreas (for example, valproic acid and diuretics), potentially significant muscle anabolic or catabolic effects or weight gain were prohibited.
Endpoints and assessments
The primary endpoint was change from baseline in lean body mass at 24 weeks as assessed by whole-body DEXA (Hologic and GE Lunar) scans in participants receiving apitegromab with tirzepatide compared to those receiving tirzepatide alone. Specifically, each DEXA scan included bilateral arms, bilateral legs and trunk (including the body organs). Cross-calibration between DEXA systems was not warranted. Secondary endpoints included change from baseline in body weight, total fat mass and DEXA parameters on body composition at week 24. Body weight was obtained using a calibrated scale in a fasted state at each visit from screening through the last trial visit. For primary and secondary endpoints based on DEXA measurements, scans were obtained within 7 days prior to participants’ initial dose of apitegromab or placebo and were repeated at 24 weeks. Exploratory endpoints included change from baseline in DEXA measurements at week 32, physical function (force production with handheld dynamometry and number of repetitions in the chair sit-to-stand test) at weeks 24 and 32 and cardiometabolic markers (for example, hemoglobin A1C (HbA1C)) at weeks 24 and 32.
Prespecified pharmacokinetics and pharmacodynamics endpoints were assessed through the treatment and safety follow-up periods for all participants who received at least one dose of apitegromab or placebo. Apitegromab and tirzepatide trough samples were collected every 4 weeks during the treatment period and during safety follow-up visits, and end-of-infusion samples of apitegromab were collected on day 1, week 12 and week 20. Trough concentrations of latent myostatin were collected through 24 weeks and during the safety follow-up period.
Safety endpoints included the frequency of AEs, clinical laboratory tests, vital signs, electrocardiogram measurements and psychiatric evaluations reported through the last study visit. Data were reviewed throughout the study in a blinded manner by medical monitors and the sponsor to ensure participant safety. The presence of serum ADAs against apitegromab was also assessed. Safety data were collected through week 40 of the study.
Statistical analysis
The primary population for efficacy analyses consisted of all randomized participants who completed treatment and had evaluable DEXA lean body mass data at baseline and at the scheduled week 24 visit (designated as the completer population in the Statistical Analysis Plan). Most efficacy analyses were repeated on the ITT population (all randomized participants) and the modified ITT population (all randomized participants with evaluable baseline and one postbaseline lean body mass measurement, regardless of timing). Safety analyses included all dosed participants, and pharmacokinetics/pharmacodynamics analyses included all participants with quantifiable pharmacokinetics/pharmacodynamics data. Analyses were conducted using SAS version 9.4 software.
All efficacy, safety and pharmacokinetics/pharmacodynamics endpoints are summarized descriptively by treatment group. Least square means together with 80% CIs for each treatment group and the difference between groups in change from baseline at week 24 in lean body mass were estimated using a linear regression model controlling for baseline weight, baseline lean body mass, age and sex. The same regression model was used to estimate least square means and differences between groups with their associated CIs for the changes in other DEXA parameters and the change in weight. Post hoc analyses for the primary endpoint included a subgroup analysis by sex and a sensitivity analysis exploring effects of study sites.
A two-sided significance level of 0.20 was selected for this phase 2 study investigating the effects of apitegromab in this population. All P values are nominal with no adjustment for multiplicity, and the CIs presented should be interpreted within this context.
Assuming a standard deviation of 4.6 kg, an evaluable sample size of 43 participants per study arm was determined to yield approximately 75% power to detect an effect size of 2 kg for the primary endpoint of lean body mass change from baseline at week 24. Anticipating that approximately 13% would not be evaluable for the primary analysis, a total of 50 participants per arm was planned.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.




